Movement of unchanged form of drug molecules from site of administration to systemic circulation is called absorption.
Various factors affect the rate of absorption they are listed as follows,
Physico-chemical,
Pharmaceutical and,
Physiological.
PHYSICOCHEMICAL FACTORS:
Drug solubility and dissolution rate
Particle size and effective surface area
Polymorphism and amorphism
Pseudo polymorphism (hydrates / solvates).
Salt form of the drug.
Lipophilicity of the drug – (pH partition hypothesis).
pKa of the drug and pH – (pH partition hypothesis).
Drug stability.
PHARMACEUTICAL FACTORS:
Disintegration time (tablets / capsules).
Dissolution time.
Manufacturing variables.
Pharmaceutical ingredients (excipients / adjuvants).
Nature and type of dosage form.
Product age and storage conditions
PHYSIOLOGICAL FACTORS:
Age.
Gastric emptying time.
Intestinal transit time.
Gastrointestinal pH.
Disease states.
Blood flow through the GIT.
Gastrointestinal contents:
Other drugs b) Food c) Fluids d) Other normal GI contents
Pre-systemic metabolism by:a) Luminal enzymes b) Gut wall enzymes c) Bacterial enzymes d) Hepatic enzymes.
PHYSICOCHEMICAL FACTORS:
Drug solubility and dissolution rate:
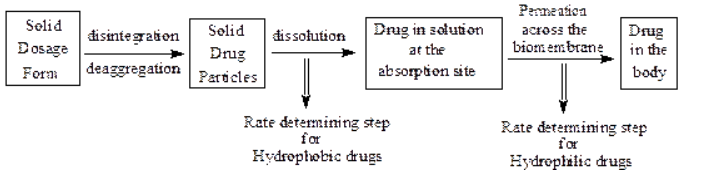
Orally administered solid dosage forms are first disintegrated or disaggregated, then the solid particles are dissolved; drugs in solution then permeate across biomembranes to be absorbed in the body.
Two critical processes in the absorption of orally administered drugs are:
Rate of dissolution, and
Rate of drug permeation through the bio membrane (i.e. gastrointestinal membrane)
For poorly water-soluble drugs rate of dissolution is the rate determining step hence the absorption is called to be dissolution rate limited. e.g. Griseofulvin, Spironolactone.
For highly water-soluble drugs dissolution is rapid so the rate determining step is permeation hence, the absorption is called to be permeation rate limited. e.g., Cromlyon sodium, Neomycin sulfate etc.
Particle size and effective surface area of the drug particles:
From Noyes-Whitney’s equation of dissolution:
where,
D = diffusion coefficient or diffusivity of the drug molecule
A = surface area of the dissolving solid exposed to the dissolution medium
Kw/o = water/oil partition coefficient of the drug
V = volume of dissolution medium
h = thickness of the stagnant layer
Cs – CB = concentration gradient of the diffusing drug molecule.
From this equation it can be concluded that the greater the surface area, A, faster the distribution.
Two types of surface area can be defined:
1. Absolute surface area: Which is the total area of solid surface of any particle, and
2. Effective surface area: Which is the area of solid surface exposed to the dissolution medium.
When the particle size of a certain mass of a drug is reduced the surface area is increased, hence, if particle size is reduced dissolution rate increases.
e.g. Micronization of poorly water soluble drugs like Griseofulvin, Chloramphenicol and several salts of Tetracycline results in superior dissolution rates.
However, size reduction has some limitations.
In the case of hydrophobic drugs like Aspirin, Phenacetin and Phenobarbital micronization actually results in a decrease in effective surface area due to the following reasons.
(i) The hydrophobic surface of the drugs adsorb air onto their surface which inhibits their wettability, such powders float on the dissolution medium.
(ii) The particles reaggregate to form larger particles due to their high surface free energy.
(iii) Extreme particle size reduction may impart surface charges that may prevent wetting and intimate contact of the drug with the dissolution medium.

Polymorphism and amorphism:
Depending on the internal structure, a solid can exist either in a crystalline or amorphous form.
When a substance exists in more than one crystalline form, the different forms are designated as polymorphs and the phenomenon as polymorphism.
Depending on their relative stability, one of the several polymorphic forms will be physically more stable than the others.
Such a stable polymorph represents the lowest energy state, has the highest melting point and least aqueous solubility.
The remaining polymorphs are called metastable forms which represent higher energy states, the metastable forms have a thermodynamic tendency to convert to the stable form.
A metastable form cannot be called unstable because if it is kept dry, it will remain stable for years.
So the metastable forms have higher aqueous solubility and hence higher bioavailability than the stable polymorphs.
e.g. Chloramphenicol palmitate has three polymorphs A, B and C. The B -form shows best bioavailability and A form is virtually inactive biologically.
e.g. Polymorphic form-III of Riboflavin is 20 times more water soluble than the form-I.
Due to aging of dosage forms containing metastable forms of the drug results in the formation of less soluble, stable polymorph.
e.g. more soluble crystalline form-III of Cortisone acetate converts to less soluble form-V in an aqueous suspension resulting in caking of solid.
Amorphous form (i.e. having no internal structure):
Such drugs represent the highest energy state and can be considered as super cooled liquids.
They have greater aqueous solubility than their crystalline form. (Less energy taken while dissolving than crystalline form as they lack definite structure.)
e.g. the amorphous form of the Novobiocin is 10 times more soluble than the crystalline form.
Thus the order for dissolution of different solid forms of drug is amorphous > metastable > stable.
Pseudo polymorphism (Hydrates / Solvates):
During the crystallization process the solvent molecules may be incorporated into the crystal lattice of the solid in stoichiometric proportion – these types of crystals are called solvates; and the trapped solvent molecules as solvent of crystallization.
The solvates again can remain in different polymorphic states, called as pseudopolymorphs. The phenomenon is called pseudopolymorphism.
When the solvent with the drug is water, the solvate is known as hydrate.
Effect on absorption:
Generally, the anhydrous form of a drug has greater solubility than the hydrates.
This is because the hydrates are already in equilibrium with water and therefore have less demand for water.
e.g. anhydrous form of Theophylline and Ampicillin have higher aqueous solubility, dissolve at faster rate and show better bioavailability in comparison to their monohydrates and trihydrate forms respectively.
On the other hand non aqueous solvates have greater aqueous solubility than the nonsolvates.
e.g. n-pentanol solvate of Fludrocortisone and Succinyl Sulfathiazole and the chloroform solvates of Griseofulvin are more water soluble than their non-solvate forms.
Salt form of the drug:
Most drugs are either weak acids or weak bases.
One of the easiest approaches to enhance the solubility and dissolution rate of such drugs is to convert them into their salt forms.
Some time in-situ salt formation can be utilized, e.g. certain drugs like aspirin and penicillin are prepared as buffered alkaline tablets.
When the tablets are put into water the pH of the microenvironment of the drug is increased which promotes the dissolution rate.
So buffered aspirin tablets have three advantages
(i) the gastric irritation and ulcerogenic tendency of the drug is greatly reduced and,
(ii) in dry form the hydrolytic stability is better.
(iii) bioavailability is increased by increasing the dissolution.
pKa of the drug and pH:
Drug pKa and lipophilicity and GI pH (pH partition theory):
The pH partition theory (Brodie et.al.) states that for drug compounds of molecular weight greater than 100, which are primarily transported across the biomembrane by passive diffusion.
The process of absorption is governed by,
1. dissociation constant (Ka) of the drug
2. lipid solubility of the unionized drug (Ko/w)
3. the pH at the absorption site
It is the pKa of the drug that determines the degree of ionization at a particular pH and that only the unionized drug, if sufficiently lipid soluble, is absorbed into the systemic circulation.
Ideally, for optimum absorption, a drug should have sufficient aqueous solubility to dissolve in the fluids at the absorption site and lipid solubility (Ko/w) in the lipoidal biomembrane and into the systemic circulation.
In other words, a perfect hydrophilic-lipophilic balance (HLB) should be there in the structure of the drug for optimum bioavailability.
B. PHARMACEUTICAL FACTORS.
Disintegration time:
Disintegration time (DT) is of particular importance in case of solid dosage forms like tablets and capsules.
After disintegration of a solid dosage form into granules, the granules must deaggregate into finer particles and then dissolution takes place.
If DT is long the bioavailability will be less.
Rapid disintegration is thus important in the therapeutic success of a solid dosage form.
DT increases with increase in the amount of binder and hardness of a tablet.
Disintegration can be made faster by incorporating disintegrants in suitable amounts during formulation.
Manufacturing / process variables:
Dissolution from a solid dosage form depends on:
(A) excipients and
(B) manufacturing process.
A) Excipients:
A drug is rarely administered in its original form.
All dosage forms contain a number of suitable excipients (non-drug components of a formulation).
1) Vehicle:
Vehicle or solvent system that carries a drug is the major component of liquid orals and parenterals.
The three categories of vehicles generally used are:
(i) aqueous vehicles e.g. water, syrup etc.
(ii) nonaqueous but water miscible e.g. propylene glycol, glycerol, sorbitol.
(iii) nonaqueous and water immiscible vehicle e.g. vegetable oils.
Bioavailability of a drug from a vehicle depends, to a large extent, on its miscibility with biological fluids.
Aqueous and water miscible vehicles are rapidly miscible with body fluids (e.g. G.I.-fluid, tissue fluid, blood etc.) and drugs are rapidly absorbed from them.
Propylene glycol, glycerol, alcohol etc. are used as co-solvent to increase the solubility of a drug in water. Sometimes solubilizers, such as Tween 80 are used to promote solubility of a drug in aqueous vehicle.
In case of water immiscible vehicles, the rate of drug absorption depends upon its partitioning from the oil phase to the aqueous body fluids, which could be a rate limiting step.
2) Diluents (Fillers):
Diluents are commonly added to tablet (and capsules) formulations.
Hydrophilic powders used as diluent are starch, lactose, microcrystalline cellulose etc. These hydrophilic powders form a coating over the hydrophobic drugs particles (e.g. spironolactone and triamterene) and render them hydrophilic.
Inorganic diluents like dicalcium phosphate (DCP) forms a divalent calcium-tetracycline complex which is poorly soluble in water and thus unabsorbable.
3) Binders and granulating agents:
These materials are used to hold powders together to form granules or promote cohesive compacts for directly compressible materials and ensure that the tablet remains intact after compression.
Large amounts of binders increase hardness and thus decrease disintegration / dissolution rates of tablets.
Non-aqueous binders like ethyl cellulose also retard dissolution.
4) Disintegrants:
These agents overcome the cohesive strength of tablets and break them up on contact with water.
Almost all the disintegrants are hydrophilic in nature.
A decrease in the amount of disintegrant can significantly lower the bioavailability.
5) Lubricants:
These agents are added to tablet formulations to aid flow granules, to reduce interparticular friction and to reduce sticking or adhesion of particles to dies and punches.
The commonly used lubricants are hydrophobic in nature e.g Magnesium stearate. They reduce the wettability of the particle surface, penetration of water into the tablet.
The best alternative is to use soluble lubricants like sodium lauryl sulfate and carbowax which promotes drug dissolution.
6) Suspending agents /Viscosity building agents:
Agents like vegetable gums (acacia, tragacanth etc.), semisynthetic gums (carboxymethyl cellulose, methyl cellulose) and synthetic gums are popular suspending agents.
The macromolecular gums often form an unabsorbable complex with amphetamine.
An increase in viscosity by these agents acts as a mechanical barrier to the diffusion of drug from the dosage form into the bulk of GI fluids.
7) Surfactants:
Surfactants are widely used in formulations as wetting agents, solubilizers, emulsifiers, etc.
Surfactants increase the absorption of a drug by the following ways:
1. Promotion of wetting (through increase in effective surface area) and dissolution of drugs e.g. Tween80 with phenacetin.
2. Better membrane contact of the drug for absorption
3. Enhanced membrane permeability of the drug .
Decreased absorption of drug in the presence of surfactants has been suggested to be due to :
1. Formation of unabsorbable drug-micelle complex at surfactant concentrations above critical micelle concentration.
2. Laxative action induced by a large surfactant concentration.
B) Manufacturing process:
1) Method of granulation:
The wet granulation process is the most conventional technique of manufacturing tablet granules. The limitation of this method include –
(i) formation of crystal bridge due to the presence of solvent,
(ii) the liquid may act as medium or affecting chemical reactions such as hydrolysis, and
(iii) the drying step may harm the thermolabile drugs.
Wet granulation includes a greater number of steps than dry granulation or direct compression which can adversely affect the dissolution.
2) Compression force:
The compression force employed in the tableting process influences density, porosity, hardness, disintegration time and dissolution of tablets.
The curve obtained by plotting compression force versus rate of dissolution can take one of the 4 possible shapes shown in the figures:
A. High compression force
↑density and hardness of tablet
↓porosity, hence penetrability of the solvent into the tablet
↓wettability by forming a firmer and more effective sealing layer by the lubricant
B. Higher compression force
causes deformation, crushing or fracture of drug particles into smaller ones or, convert a spherical granules into a disc shaped particle with large increase in effective surface area
↑ in dissolution rate
C and D are combinations of both the causes of A and B.
In short, the influence of compression force on the dissolution is difficult to predict.
3) Intensity of packing of capsule contents:
Packing density in the case of a capsule can either inhibit or promote dissolution.
Diffusion of GI fluids into the tightly filled capsules creates a high pressure within the capsule resulting in rapid bursting and dissolution of contents.
In some cases capsules with tight packing
pore size of the compact mass is decreased
poor penetrability of GI - fluid
poor rate of drug release.
4) NATURE AND TYPES OF DOSAGE FORM:
As a general rule, the bioavailability of a drug from various dosage forms decreases in the following order:
Solution > Emulsions > Suspensions > Capsules > Tablets > Coated tablets > Enteric coated tablets > Sustained release tablets.
Thus, absorption of a drug from solution is fastest with least potential for bioavailability problems whereas absorption from sustained release product is lowest with greatest bioavailability.

C) Physiological Factors / Patient Related Factors:
Age:
In infants,
gastric: pH is high
intestinal surface is small
blood flow is less.
In elderly persons:
altered gastric emptying
decreased intestinal surface area
decreased GI blood flow
achlorhydria
bacterial overgrowth in the small intestine.
In both of these ages drug absorption is impaired.
Gastric emptying:
Passage of gastric content from stomach to small intestine is called gastric emptying.
Rapid gastric emptying is required where:
(i) a rapid onset of action is required e.g. Sedatives.
(ii) dissolution of drug occurs in the intestine e.g. enteric coated dosage forms.
(iii) the drugs are not stable in gastric fluid e.g. Penicillin-G and Erythromycin.
(iv) the drug is best absorbed from the distal part of the small intestine e.g. vitamin B12.
Delay in gastric emptying is required where:
(i) the food promotes drug dissolution and absorption e.g. Griseofulvin
(ii) disintegration and dissolution of dosage form is promoted by gastric fluid
(iii) the drugs are absorbed from the proximal part of the small intestine e.g. vitamin B2 and vitamin C.
Gastric emptying is a first order rate process. Several parameters are used to quantify gastric emptying:
(i) Gastric emptying rate is the rate at which the stomach contents empty into the intestine.
(ii) Gastric emptying time is the time required for the gastric content to empty completely into the small intestine.
(iii) Gastric emptying t1/2 is the time taken for half the stomach contents to empty.
N.B. In vivo gastric emptying can be studied by using radio-opaque contrast materials (e.g. BaSO4) or tagging the drug with a radio-isotope and scanning the stomach at regular intervals of time.
Factors influencing gastric emptying rate:-
1) Volume of meal:
Larger the volume of the meal, the longer the gastric emptying time.
2) Composition of meal:
The rate of gastric emptying for various food materials is in the following order:
carbohydrates > protein > fats.
3) Physical state and viscosity of meal:
Liquid meals take less than an hour to empty solid meals take as long as 6 – 7 hours to empty.
Viscous material empty at a slow rate in comparison to less viscous materials.
4) Temperature of the meal:
High or low temperature of the ingested fluid (compared to body temperature) reduces gastric emptying rate.
5) Gastrointestinal pH:
Gastric emptying is retarded at low stomach pH and is promoted at higher or alkaline pH.
6) Electrolyte and osmotic pressure:
Water, isotonic, and solutions of low salt concentration empty the stomach rapidly whereas higher electrolyte concentration decreases gastric emptying rate.
7) Body posture:
Gastric emptying is favored while standing and while lying on the right side; while lying on the left side or in supine position retards it.
8) Emotional state:
Stress and anxiety promote gastric motility whereas depression retards it.
9. Exercise
Vigorous physical exercise retards gastric emptying.
10 Disease states
Diseases like gastroenteritis, gastric ulcer, pyloric stenosis, diabetes and hypothyroidism retard gastric emptying.
11. Drugs
Drugs that retard gastric emptying includes
(i) poorly soluble antacids e.g. Aluminum hydroxide,
(ii) Anticholinergics e.g. Atropine, Propantheline
(iii) Narcotic analgesics e.g. Morphine
(iv) Tricyclic antidepressants e.g. Imipramine, Amitriptyline.
Drug that stimulate gastric emptying are:
(i) Metoclopramide (Metopar)
(ii) Domperidone (Domstal)
(iii) Cisapride (Alpride)
Effect of GI pH on drug absorption:
GI fluid pH influence drug absorption in several ways:
1. Disintegration:
The disintegration of some dosage forms is pH sensitive. With enteric coated formulations, the coat dissolves only in the intestinal pH, followed by disintegration of the tablet.
2. Dissolution:
A large number of drugs are either weakly acidic or weakly basic whose solubility is greatly affected by pH. A pH that favors the formation of salt of the drug enhances the dissolution rate. e.g. Weakly acidic drugs dissolve rapidly in the alkaline pH of the intestine whereas basic drugs dissolve in the acidic pH of the stomach.
3. Absorption:
Depending upon the pKa of the drug and the pH of the GI fluid some amount of the drug remains in ionized state and some in unionized state.
The unionized form will be absorbed through GIT more quickly than the ionized form.
4. Stability:
GI pH influences the chemical stability of drugs. e.g. The acidic stomach pH is known to affect degradation of Penicillin-G and Erythromycin.
Effect of GI content:
Presence of food may either delay, reduce, increase or may not affect drug absorption.
As a general rule, drugs are better absorbed under fasting conditions and presence of food retards or prevents it.
Food does not significantly influence absorption of a drug taken half an hour or more before meals and two hours or more after meals.
Delayed or decrease drug absorption by food can be due to one or more of the following reasons:
(a) Delayed gastric emptying, affecting the drugs unstable in the stomach e.g. penicillin, erythromycin.
(b) Preventing the transit of enteric tablets into the intestine which may be as long as 6 – 8 hrs.
(c) Formation of poorly soluble, unabsorbable complex e.g. Tetracycline-Calcium complex (Chelate).
(d) Increased viscosity due to food thereby preventing drug dissolution and/or diffusion towards the absorption site.
Increased drug absorption following a meal can be due to the following reasons:
(a) Increased time for dissolution of poorly soluble drug.
(b) Enhanced solubility due to GI secretions like bile.
(c) Prolonged residence time and absorption site contact of the drug e.g. water-soluble vitamins.
Types of meal:
(i) Meals high in fat aid solubilisation of poorly aqueous soluble drugs like Griseofulvin.
(ii) Food high in proteins increases oral availability of propranolol because
a) such a meal promotes blood flow to the GIT helping in drug absorption.
b) increases hepatic blood flow due to which the drug can bypass first-pass hepatic metabolism (Propranolol is a drug with high hepatic metabolism).

Drug-drug interaction:
Drug-drug interactions can be either physicochemical or physiological.
(a) Physicochemical drug-drug interactions can be due to –
1) Adsorption:
Antidiarrheal preparations containing adsorbents like attapulgite or kaolin-pectin retard / inhibit absorption of Promazine and Lincomycin when co-administered with them.
2) Complexation:
Antacids containing heavy metals such as Aluminium, Calcium, Iron, Magnesium or Zinc retard absorption of tetracycline due to the formation of unabsorbable complexes called as chelates.
3) pH change:
Basic drugs dissolve in gastric pH. Co-administration of sodium bicarbonate with tetracycline results in evaluation of stomach pH and hence decreases dissolution rate or causes precipitation of drug.
(b) Physiologic drug-drug interaction can be due to following reasons:
1) Decreased GI transit:
Anticholinergic drugs such as propantheline retard GI motility and promote absorption of drugs like ranitidine and digoxin.
2) Increased gastric emptying:
Metoclopramide promotes GI motility and enhances absorption of Tetracycline, Pivampicillin and Levodopa.
3) Altered GI metabolism:
Antibiotics inhibit bacterial metabolism of drugs e.g. Erythromycin enhances the efficacy of digoxin by this mechanism.
Presystemic metabolism / First pass effects:
The loss of drug through biotransformation by GIT and liver during the passage to systemic circulation is called First pass or presystemic metabolism.
The 4 primary systems which affect presystemic metabolism of drugs are:
1. Luminal enzymes
2. Gut wall enzymes /mucosal enzymes
3. Bacterial enzymes, and
4. Hepatic enzymes
1. Luminal enzymes
These are enzymes present in the gut fluids and include enzymes from intestinal and pancreatic secretions.
Pancreatic enzymes contain hydrolases which hydrolyze ester drugs like chloramphenicol palmitate into active chloramphenicol.
Peptidases split amide ( –CONH) linkages and inactivate protein / polypeptide drugs. Thus one of the approaches is to deliver them to a colon which lacks peptidases.
2. Gut-wall enzymes (also called mucosal enzymes):
They are present in the stomach, intestine and colon.
Stomach mucosa contains alcohol dehydrogenase (ADH) inactivates ethanol.
3. Bacterial enzymes:
The GI microorganisms are scantily present in the stomach and small intestine and are rich in the colon. Hence, most orally administered drugs remain unaffected by them.
The colonic microbes generally render a drug more active or toxic on biotransformation:
e.g. Sulfasalazine (used in ulcerative colitis) is hydrolyzed to Sulfapyridine and 5-amino salicylic acid by the microbial enzymes of the colon.
Digoxin, oral contraceptive drugs are absorbed in the upper intestine; exerted through bile as glucuronide conjugates.
These conjugates of drugs are hydrolyzed by microbial enzymes.
The free drugs are reabsorbed into the systemic circulation.
4. Hepatic enzymes:
e.g. Isoprenaline, Propranolol, Alprenolol, Pentoxifylline, Nitroglycerin, Diltiazem, Nifedipine, lidocaine, morphine etc.
Commonly Asked Question.
Discuss in detail factors affecting absorption of drugs.